Multivariate O-statistics
Quentin D. Read, Isadora E. Fluck
Source:vignettes/Ostats-multivariate.Rmd
Ostats-multivariate.RmdOverview
This vignette on multi-variable community-level trait overlap statistics is intended for users who are already familiar with the single-variable O-statistics we introduce in the first package vignette.
We decided to include functions in our package to estimate community-level niche overlap statistics (O-statistics) in multivariate space because of ecologists’ long-standing interest in how species’ niches take shape across multiple environmental dimensions. Since the niche of a species can be determined by several abiotic and biotic variables, Hutchinson (1957) introduced the mathematical formulation of a niche as an n-dimensional hypervolume. Since then, many approaches have been developed to measure the multivariate niche of a species (Green 1971; Swanson et al. 2015). Quantifying the niche in multiple dimensions can help understand how life strategy trade-offs and interactions among multiple traits shape organisms’ structure and function.
Blonder and colleagues rekindled interest in using hypervolumes to
describe species’ niches in multiple dimensions. They worked out the
underlying theory (Blonder et al. 2014; Lamanna et al. 2014; Blonder
2018) and released the R package hypervolume.
Their package uses stochastic geometry to estimate multivariate kernel
densities and . The multivariate O-statistic estimation in the
Ostats package relies on functions imported from hypervolume.
In this vignette, we demonstrate how you can use functions in the
Ostats package to study trait overlap in multiple
dimensions.
Example dataset
The dataset we use in this vignette includes traits collected on
carnivorous pitchers of the northern pitcher plant, Sarracenia
purpurea, at 5 sites in North America and represent a subset of
data from Freedman et al. (2021). Pitcher traits measured include
orthogonal rosette diameters, pitcher length, pitcher width, keel width,
mouth diameter, and lip thickness. See Ellison and Gotelli (2002) for a
diagram illustrating traits measured, or see the dataset documentation
page for more details by typing ?pitcher_traits in your
console. Site locations and identifying information have been removed
for this example.
Here we show a few randomly selected rows from the pitcher traits
dataset. Here, all individuals are of the same species. Therefore we
will use populations (sites) as the sp variable in our
analysis, with a single value for plots.
set.seed(1)
idx <- sort(sample(nrow(pitcher_traits), 10, replace = FALSE))
knitr::kable(pitcher_traits[idx,])| site_id | plant_id | rosette_diameter_1 | rosette_diameter_2 | pitcher_length | pitcher_width | keel_width | mouth_diameter | lip_thickness | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | FLK | 1 | 16 | 14.0 | 15.4 | 42.40 | 13.25 | 29.31 | 4.48 |
| 14 | FLK | 14 | 13 | 3.0 | 9.7 | 30.63 | 9.77 | 20.05 | 3.05 |
| 60 | MYR | 10 | 20 | 10.0 | 15.0 | 44.88 | 12.75 | 26.67 | 4.04 |
| 65 | MYR | 15 | 29 | 16.0 | 16.2 | 48.28 | 19.74 | 22.09 | 3.13 |
| 69 | MYR | 19 | 5 | 9.0 | 17.9 | 50.55 | 17.62 | 24.50 | 3.27 |
| 203 | CRI | 3 | 32 | 17.0 | 21.5 | 32.79 | 11.07 | 21.49 | 2.39 |
| 211 | CRI | 11 | 11 | 9.5 | 17.5 | 37.42 | 16.48 | 20.73 | 1.87 |
| 220 | CRI | 20 | 52 | 31.0 | 19.0 | 38.00 | 13.78 | 25.13 | 2.57 |
| 282 | SAC | 10 | 11 | 8.5 | 15.5 | 42.05 | 17.60 | 21.55 | 2.90 |
| 287 | SAC | 15 | 23 | 23.0 | 13.0 | 34.77 | 12.75 | 22.15 | 1.11 |
Calculating hypervolume O-statistics
Here we calculate the univariate O-statistics for each of the pitcher traits separately, then a single multivariate O-statistic representing overlap of the trait hypervolumes.
When estimating hypervolumes, as with any other multivariate analysis, it is critical to ensure that the hypervolume axes are not too highly correlated with one another. If traits are too highly correlated, the calculated hypervolume will not accurately represent the distribution in trait space. In this case, we resolve the issue by excluding a few of the traits with high correlations. You could also choose to use a variance maximization procedure like PCA instead.
First ensure that the data are in the appropriate form for
Ostats() and Ostats_multivariate(). For this
example we will remove individuals with incomplete data. Using
cor() on the cleaned dataset, we can see the relatively
high correlations among some of the traits.
pitcher_traits <- pitcher_traits[complete.cases(pitcher_traits), ]
pitcher_sites <- as.factor(pitcher_traits$site_id)
round(cor(pitcher_traits[, -(1:2)]), 2)
#> rosette_diameter_1 rosette_diameter_2 pitcher_length
#> rosette_diameter_1 1.00 0.83 0.64
#> rosette_diameter_2 0.83 1.00 0.71
#> pitcher_length 0.64 0.71 1.00
#> pitcher_width 0.38 0.44 0.70
#> keel_width 0.46 0.51 0.69
#> mouth_diameter 0.38 0.35 0.43
#> lip_thickness -0.11 -0.19 -0.15
#> pitcher_width keel_width mouth_diameter lip_thickness
#> rosette_diameter_1 0.38 0.46 0.38 -0.11
#> rosette_diameter_2 0.44 0.51 0.35 -0.19
#> pitcher_length 0.70 0.69 0.43 -0.15
#> pitcher_width 1.00 0.82 0.49 0.12
#> keel_width 0.82 1.00 0.29 -0.03
#> mouth_diameter 0.49 0.29 1.00 0.47
#> lip_thickness 0.12 -0.03 0.47 1.00We select four traits that represent dimensions of different parts of the plant’s anatomy. As a result they are relatively uncorrelated with one another: rosette diameter, pitcher width, mouth diameter, and lip thickness.
traits_to_use <- c("rosette_diameter_1", "pitcher_width", "mouth_diameter", "lip_thickness")
pitcher_trait_matrix <- as.matrix(pitcher_traits[, traits_to_use])
round(cor(pitcher_trait_matrix), 2)
#> rosette_diameter_1 pitcher_width mouth_diameter
#> rosette_diameter_1 1.00 0.38 0.38
#> pitcher_width 0.38 1.00 0.49
#> mouth_diameter 0.38 0.49 1.00
#> lip_thickness -0.11 0.12 0.47
#> lip_thickness
#> rosette_diameter_1 -0.11
#> pitcher_width 0.12
#> mouth_diameter 0.47
#> lip_thickness 1.00Next we center and scale all the columns to ensure that the hypervolume O-statistic is not influenced by different magnitudes of each trait.
apply(pitcher_trait_matrix, 2, range)
#> rosette_diameter_1 pitcher_width mouth_diameter lip_thickness
#> [1,] 4 22.28 9.43 0.60
#> [2,] 65 83.01 36.66 6.44
pitcher_trait_matrix_scaled <- scale(pitcher_trait_matrix, center = TRUE, scale = TRUE)
round(apply(pitcher_trait_matrix_scaled, 2, range), 2)
#> rosette_diameter_1 pitcher_width mouth_diameter lip_thickness
#> [1,] -1.31 -1.96 -2.64 -1.81
#> [2,] 3.62 4.36 2.31 3.21Univariate versus multivariate O-statistics
For demonstration purposes, let’s calculate the univariate O-statistics for each of the four traits so that we can compare them to the multivariate statistic.
pitcher_univariate <- Ostats(traits = pitcher_trait_matrix_scaled,
plots = factor(rep(1, nrow(pitcher_trait_matrix_scaled))),
sp = pitcher_sites,
run_null_model = FALSE
)Now calculate the multivariate O-statistic for the traits combined.
We still specify the argument random_seed even in the
absence of a null model, because the hypervolumes are constructed with a
stochastic algorithm.
pitcher_multivariate <- Ostats_multivariate(traits = pitcher_trait_matrix_scaled,
plots = factor(rep(1, nrow(pitcher_trait_matrix_scaled))),
sp = pitcher_sites,
random_seed = 333,
run_null_model = FALSE,
hypervolume_args = list(method = 'box'),
hypervolume_set_args = list(num.points.max = 1000)
)Notice that Ostats_multivariate() accepts additional
arguments to hypervolume::hypervolume(), which it uses to
construct the hypervolumes, and
hypervolume::hypervolume_set(), which it uses to estimate
the hypervolume overlaps. For example, the default method for
hypervolume construction is method = "gaussian", and the
default maximum number of points to use for hypervolume overlap
estimation is num.points.max = 10^(3+sqrt(n)), where
n is the number of dimensions. In this vignette, we
modified the defaults to use a different method ("box") for
hypervolume construction and fewer points (1000) for
overlap estimation:
hypervolume_args = list(method = 'box'), hypervolume_set_args = list(num.points.max = 1000).
Those options speed processing time dramatically. See the help
documentation for hypervolume::hypervolume and
hypervolume::hypervolume_set for more details.
Compare the univariate and multivariate O-statistics.
pitcher_univariate$overlaps_norm
#> rosette_diameter_1 pitcher_width mouth_diameter lip_thickness
#> 1 0.528308 0.7419213 0.8198156 0.5733967
pitcher_multivariate$overlaps_norm
#> [,1]
#> 1 0.7784172The single multivariate O-statistic is not comparable to the univariate statistics because of differing dimensionality. Therefore we need to evaluate them against null models.
Null models for multivariate O-statistics
If run_null_model is set to TRUE,
Ostats_multivariate() will do nperm iterations
of a null model, defaulting to 99. Here we use a smaller
number for demonstration purposes.
First, for comparison purposes, run the null models for the univariate O-statistics for each of the four traits.
pitcher_univariate_withnull <- Ostats(traits = pitcher_trait_matrix_scaled,
plots = factor(rep(1, nrow(pitcher_trait_matrix_scaled))),
sp = pitcher_sites,
random_seed = 666,
run_null_model = TRUE,
nperm = 50
)Next, do the same for the multivariate overlap.
pitcher_multivariate_withnull <- Ostats_multivariate(traits = pitcher_trait_matrix_scaled,
plots = factor(rep(1, nrow(pitcher_trait_matrix_scaled))),
sp = pitcher_sites,
random_seed = 555,
run_null_model = TRUE,
nperm = 50,
hypervolume_args = list(method = 'box'),
hypervolume_set_args = list(num.points.max = 1000)
)Comparing the standardized effect sizes, normalized to remove the effect of varying sample sizes across sites, we can see that three of the four individual traits have O-statistics significantly lower than the null expectation (lower than the 2.5%ile of the null distribution), while the multivariate O-statistic is not significantly different from the null expectation:
| O-statistic | rosette diameter | pitcher width | mouth diameter | lip thickness | multivariate |
|---|---|---|---|---|---|
| observed | 0.528 | 0.742 | 0.820 | 0.573 | 0.768 |
| null 2.5% | 0.722 | 0.760 | 0.767 | 0.731 | 0.766 |
| null 97.5% | 0.866 | 0.875 | 0.888 | 0.869 | 0.812 |
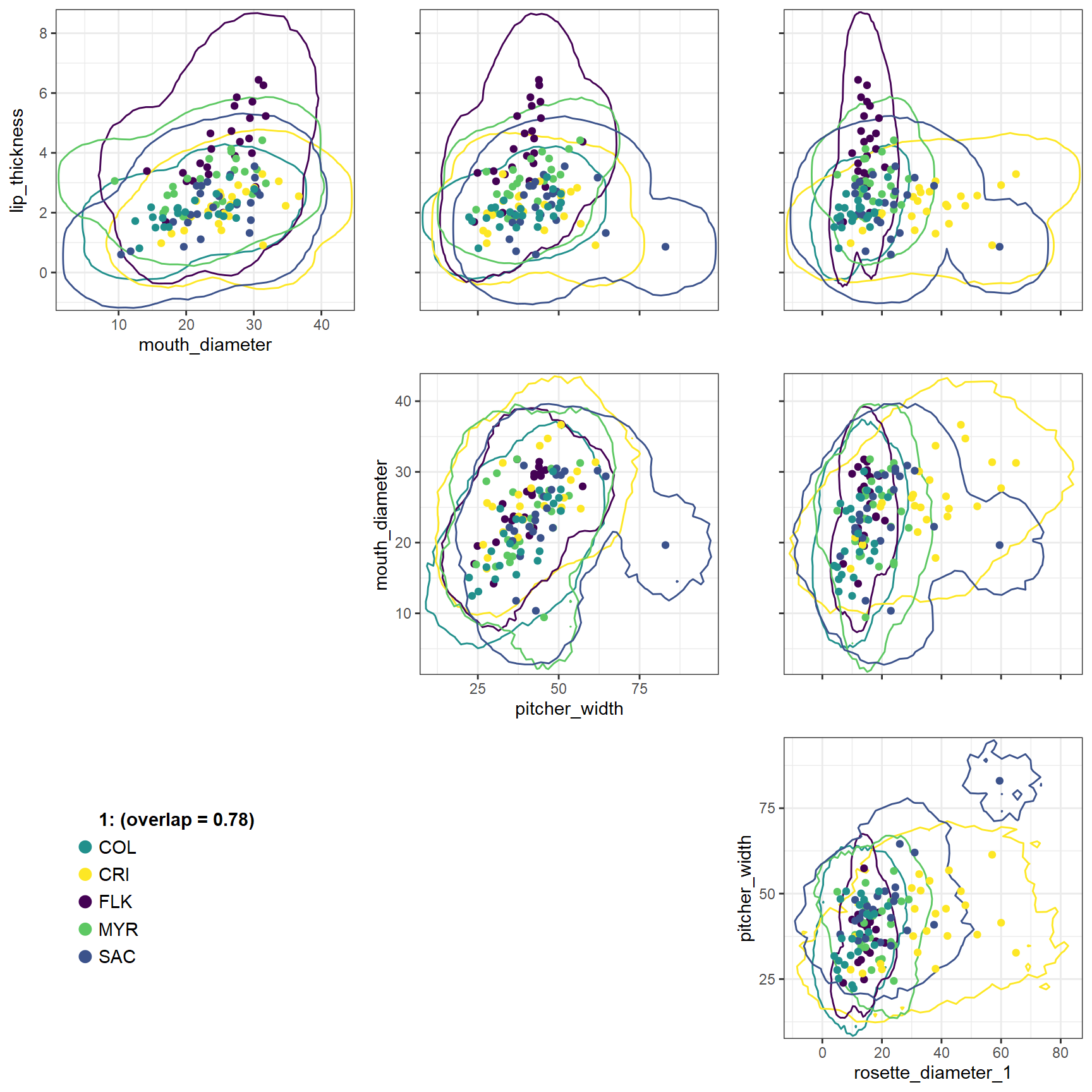
Visualizing hypervolume O-statistics
We can visualize the overlap of the different hypervolumes using the
function Ostats_multivariate_plot(). This function returns
a list of plot objects, one for each community. Each plot displays an
upper triangular matrix of panels, one for each pair of traits, with the
cross-sections of the hypervolumes in those two dimensions, as well as
the raw data points.
For example, we can plot the overlap of the hypervolumes in
4-dimensional space formed by the individual traits of the pitcher
plants at each site. Note that although we calculated the hypervolume
overlaps on the scaled traits, we are plotting the hypervolumes using
the unscaled trait axes. However, this requires us to set the argument
contour_level = 0.0001 because the densities of the
unscaled hypervolumes are too low to plot using the default
contour_level = 0.01.
Ostats_multivariate_plot(plots = factor(rep(1, nrow(pitcher_trait_matrix))),
sp = pitcher_sites,
traits = pitcher_trait_matrix,
contour_level = 0.0001,
overlap_dat = pitcher_multivariate
)
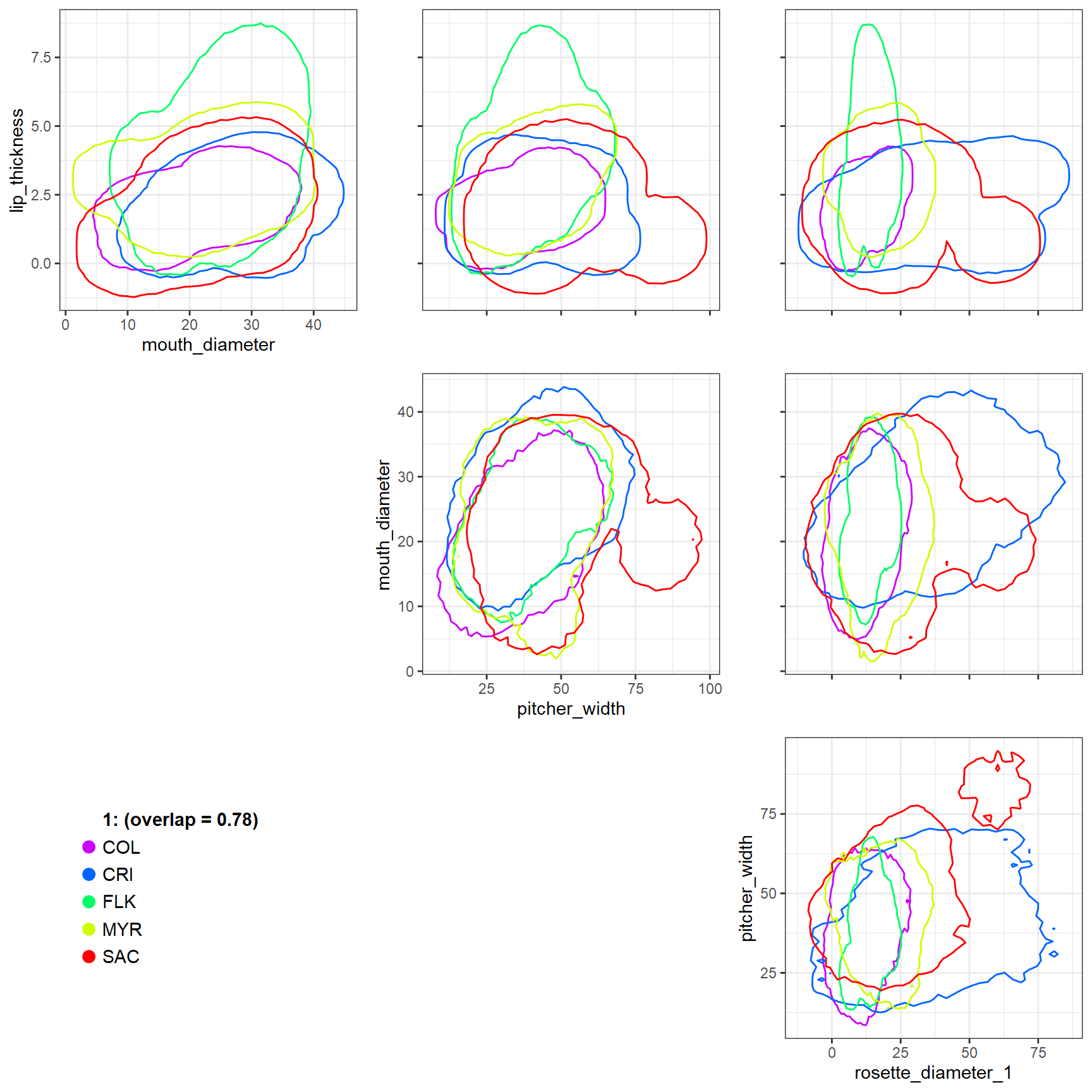
We can customize the plot by supplying a color palette to the
colorvalues argument (the default is a colorblind-friendly
palette imported from viridis),
and add additional spacing around the hypervolume contours using the
axis_expansion argument. The default value of this argument
is 0.01, or 1% in all directions. Finally, we can suppress
plotting the raw data and only plot the contours using
plot_points = FALSE.
Ostats_multivariate_plot(plots = factor(rep(1, nrow(pitcher_trait_matrix))),
sp = pitcher_sites,
traits = pitcher_trait_matrix,
contour_level = 0.0001,
overlap_dat = pitcher_multivariate,
colorvalues = rainbow(length(unique(pitcher_sites))),
axis_expansion = 0.05,
plot_points = FALSE
)
The code underlying Ostats_multivariate_plot() was
adapted from the plot.Hypervolumelist() function in the hypervolume
package.
Some practical notes
When working with multivariate trait data, it is critical to examine correlations among traits in the dataset. If you construct hypervolumes with traits that are highly correlated and measure their overlap, you will get spurious results. In addition, it’s important to consider the issue of dimensionality. It is difficult if not impossible to estimate hypervolume overlap in more than five or six dimensions. A study that quantified trait space across a wide variety of systems found that using three to six dimensions jointly maximizes trait space quality and computational feasibility (Mouillot et al., in press). Others have critiqued using hypervolume kernel density estimation to quantify species niches (Qiao et al. 2017); the technical issues only get worse as more dimensions are added. Using a variance maximization technique like PCA could be helpful to reduce dimensionality. Regardless of what method you choose, the absolute amount of overlap can’t be compared among hypervolumes with different dimensionality, making it especially important to compare the O-statistics with null distributions.
Finally, some work has shown that trait-based inference derived from analyzing multiple traits may not improve our understanding of biological systems in many cases, relative to inference based on one well-chosen biologically meaningful trait, especially when many commonly-measured traits are different manifestations of the same few underlying biological processes (Swenson 2013, but see Kraft et al. 2015). In light of that, you should carefully consider which trait or traits are relevant to your research question. Whenever possible, compare inference based on single trait overlap to the inference you get when considering multiple traits, and select the most parsimonious model.
Works cited
- Blonder, B. 2018. Hypervolume concepts in niche- and trait-based ecology. Ecography 41:1441–1455.
- Blonder, B., C. Lamanna, C. Violle, and B.J. Enquist, 2014. The n-dimensional hypervolume. Global Ecology and Biogeography 23:595–609.
- Ellison, A.M., and N.J. Gotelli. 2002. Nitrogen availability alters the expression of carnivory in the Northern Pitcher Plant, Sarracenia purpurea. Proceedings of the National Academy of Science 99(7):4409-12.
- Freedman, Z.B., A. McGrew, B. Baiser, M. Besson, D. Gravel, T. Poisot, S. Record, L.B. Trotta, and N.J. Gotelli. 2021. Environment-host-microbial interactions shape the Sarracenia purpurea microbiome at the continental scale. Ecology 102(5):e03308.
- Green, R.H. 1971. A multivariate statistical approach to the Hutchinsonian niche: bivalve molluscs of central Canada. Ecology 52:543–556.
- Hutchinson, G.E. 1957. Concluding remarks. Cold Spring Harb. Symp. Quant. Biol. 22:415–427.
- Kraft, N.J.B., O. Godoy, and J.M. Levine. 2015. Plant functional traits and the multidimensional nature of species coexistence. Proceedings of the National Academy of Sciences 112:797–802.
- Lamanna, C., B. Blonder, C. Violle, N.J.B. Kraft, B. Sandel, I. Imova, J.C. Donoghue, J.-C. Svenning, B.J. McGill, B. Boyle, V. Buzzard, S. Dolins., P.M. Jorgensen., A. Marcuse-Kubitza, N. Morueta-Holme, R.K. Peet, W.H. Piel, J. Regetz, M. Schildhauer, N. Spencer, B. Thiers, S.K. Wiser., and B.J. Enquist. 2014. Functional trait space and the latitudinal diversity gradient. Proceedings of the National Academy of Sciences 111:13745–13750.
- Mouillot, D., N. Loiseau, M. Grenié, A.C. Algar, M. Allegra, M.M. Cadotte, N. Casajus, P. Denelle, M. Guéguen, A. Maire, B. Maitner, B.J. McGill, M. McLean, N. Mouquet, F. Munoz, W. Thuiller, S. Villéger, C. Violle, and A. Auber. The dimensionality and structure of species trait spaces. Ecology Letters, in press.
- Qiao, H., L.E. Escobar, E.E. Saupe, L. Ji, and J. Soberón. 2017. A cautionary note on the use of hypervolume kernel density estimators in ecological niche modelling. Global Ecology and Biogeography 26:1066–1070.
- Swanson, H.K., M. Lysy, M. Power, A.D. Stasko, J.D. Johnson, and J.D. Reist. 2015. A new probabilistic method for quantifying n-dimensional ecological niches and niche overlap. Ecology 96:318–324.
- Swenson, N.G. 2013. The assembly of tropical tree communities – the advances and shortcomings of phylogenetic and functional trait analyses. Ecography 36:264–276.